Une étude associant les expertises en modélisation cellulaire, criblage à haut débit et analyse fonctionnelle des laboratoires I-Stem et Généthon, à Genopole, identifie deux molécules prometteuses pour le traitement de la myopathie des ceintures de type R2, pour laquelle aucune solution thérapeutique n’existe aujourd’hui.
La myopathie des ceintures de type R2 (LGMD-R2) est une maladie rare qui entraîne un affaiblissement progressif des muscles des épaules et du bassin. Bien que les caractéristiques cliniques de la LGMD-R2 soient bien décrites, les mécanismes moléculaires impliqués restent partiellement élucidés, ce qui rend difficile le développement de traitements.
La maladie est causée par des mutations du gène de la dysferline, notamment des mutations conduisant à l’absence de dysferline ou la production d’une dysferline non fonctionnelle. Or cette protéine est essentielle à la réparation de la membrane des cellules musculaires, et donc à la capacité de résistance de ces cellules aux forces mécaniques auxquelles elles sont constamment soumises.
Dans certains cas, la mutation peut entrainer la production d’une forme de dysferline fonctionnelle mais agrégée, voire dégradée par les systèmes de dégradation cellulaire. C’est le cas de la mutation L1341P qui s’exprime chez quelques patients dans le monde.
C’est sur cette mutation que les deux laboratoires génopolitains I-Stem et Généthon (AFM-Téléthon, Inserm, Université d’Évry Paris-Saclay) ont réalisé leurs travaux publiés le 20 mars dans British Journal of Pharmacology.
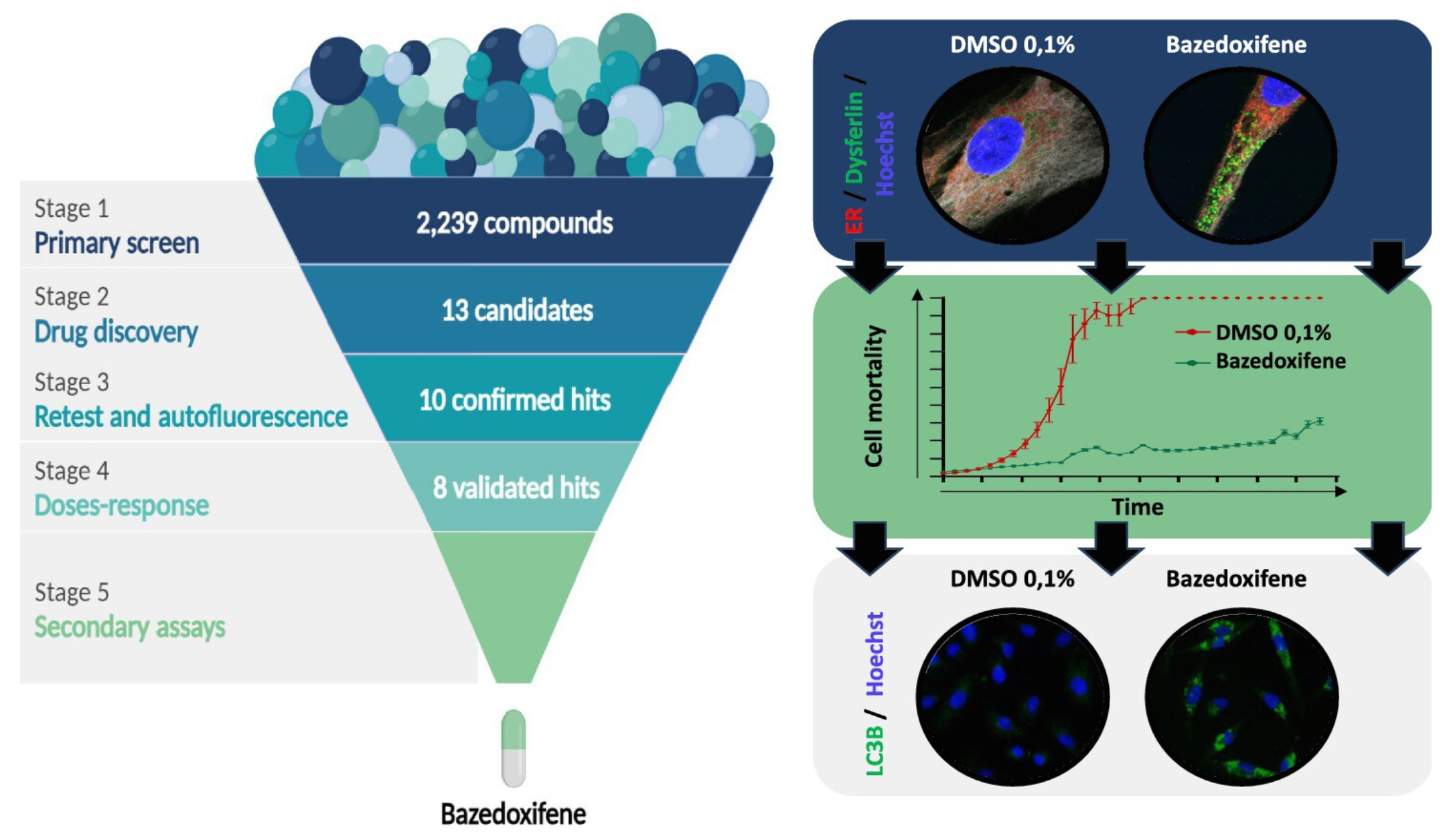
2239 molécules pharmacologiques passées au crible sur modèle cellulaire
Leur but était de trouver un médicament capable d’empêcher la dégradation de la protéine de manière à améliorer la capacité des cellules musculaires à réparer leur membrane. A cette fin, les équipes ont développé et réalisé un criblage à haut débit de médicaments sur des myoblastes immortalisés portant la mutation L1341P, préalablement mis au point par l’Institut de myologie.
Céline Bruge, chercheuse de l’équipe d’I-Stem « Pharmacologie des dystrophies musculaires » dirigée par Xavier Nissan, a d’abord reproduit l’agrégation de la dysferline et un défaut de réparation membranaire. Elle a ensuite testé une librairie de 2239 molécules, principalement composée de médicaments déjà approuvés dont le repositionnement pour la LGMD-R2 représenterait un gain de temps pour les malades.
Le saracatinib et le bazédoxifène améliorent la réparation membranaire
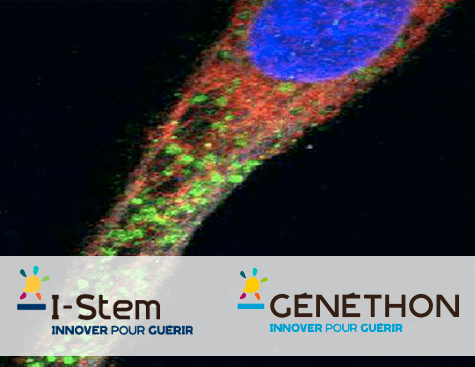
Parmi ces milliers de composés, huit candidats-médicaments ont été sélectionnés pour leur capacité à relocaliser la dysferline en dehors du reticulum endoplasmique, et ainsi éviter sa dégradation et accroître la quantité de la protéine dans la cellule (cf. photo en haut : en vert, la dysferline relocalisée dans la cellule musculaire, de forme allongée).
Un test ultérieur de la capacité à réparer la membrane plasmique après un choc hypo-osmotique, mimant un stress membranaire, a distingué deux molécules parmi les huit, le saracatinib et le bazédoxifène.
Cette caractérisation fonctionnelle a démontré l’effet protecteur des deux composés sur la membrane plasmique des cellules musculaires portant la mutation L1341P. Elle révèle également que l’effet positif sur la résistance au choc osmotique du bazédoxifène s’étend à d’autres cellules testées, y compris les cellules qui ne produisent pas de dysferline et les cellules saines, ce qui suggère un mode d’action différent des deux candidats-médicaments.
Large effet protecteur du bazédoxifène
Prenant le relais, l’équipe de Généthon « Dystrophies musculaires progressives » dirigée par Isabelle Richard, a démontré l’effet protecteur du bazédoxifène sur des fibres musculaires issues de souris déficientes en dysferline. Bien que ce médicament dispose déjà d’une AMM (autorisation de mise sur le marché) pour une autre indication thérapeutique, des essais in vivo à long terme sur des souris déficientes en dysferline seront nécessaires avant d’envisager un essai clinique